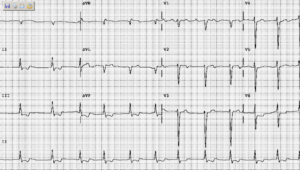
Case 6
A 35-year-old woman with Marfan syndrome presented with increasing dyspnea, fever, productive cough, and chest tightness for one day.


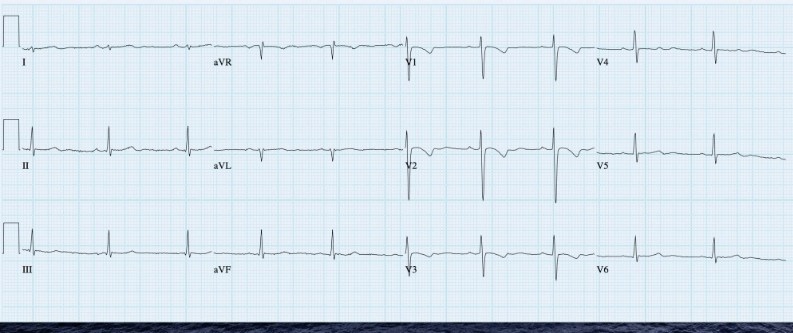
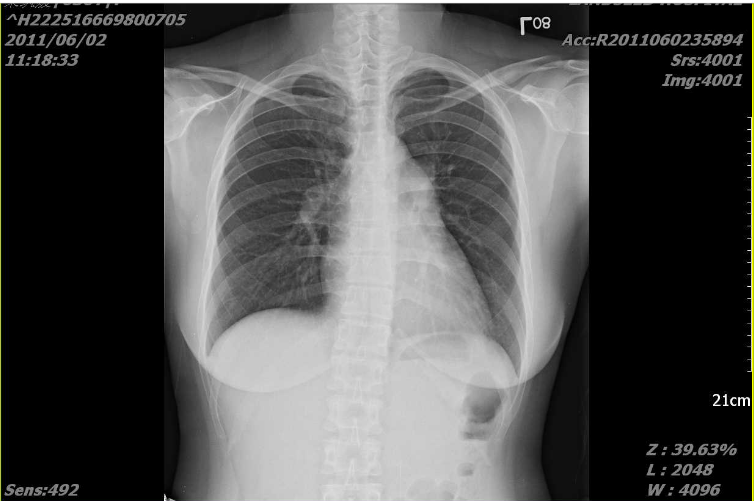
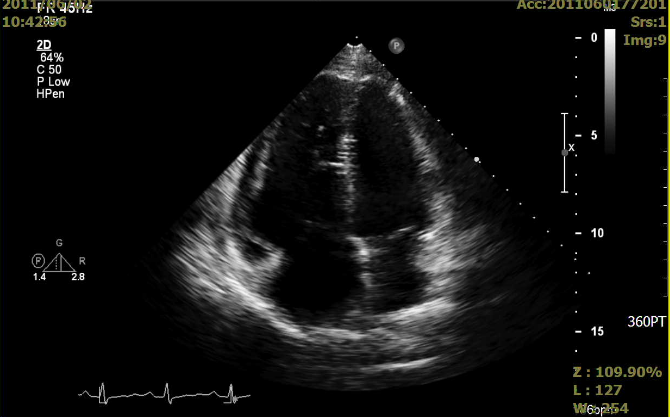
A 35-year-old woman with Marfan syndrome presented with increasing dyspnea, fever, productive cough, and chest tightness for one day.
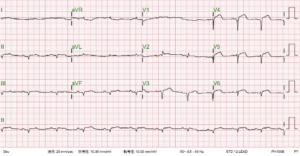
This 69-year-old man had been in good health until two days before admission when he returned from a beach. He experienced general weakness with chest
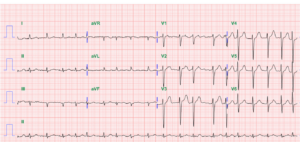
An 84-year-old man with a known history of hypertension, COPD, diabetes, Parkinsonism, and old CVA (putamen infarction)* was noted to be delirious after five days